Pytania i odpowiedzi dot. weryfikacji autentyczności leków!
26.06.2020
Tutaj znajdą Państwo odpowiedzi na najczęściej zadawane pytania dotyczące weryfikacji autentyczności leków.

- W której hurtowni farmaceutycznej uczestniczącej w łańcuchu dystrybucji produktu leczniczego powinna być wykonywana weryfikacja niepowtarzalnego identyfikatora?
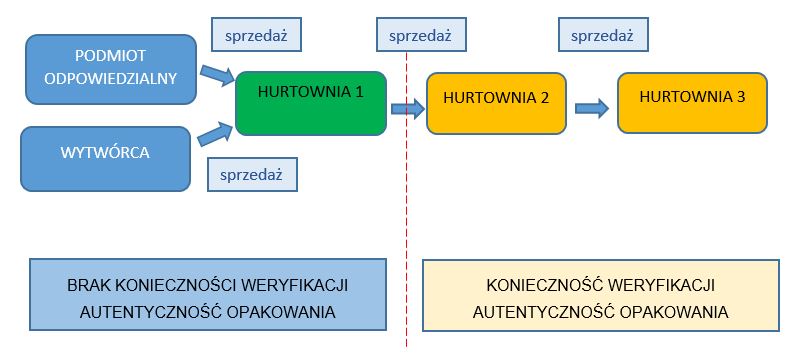
Hurtownie, które powinny dokonywać weryfikacji niepowtarzalnego identyfikatora przedstawiono na poniższych schematach:
a) Schemat bez hurtowni wskazanej w umowie o której mowa w art. 77a ustawy Prawo farmaceutyczne.
b) Schemat z uwzględnieniem hurtowni wskazanej w umowie o której mowa w art. 77a ustawy Prawo farmaceutyczne.
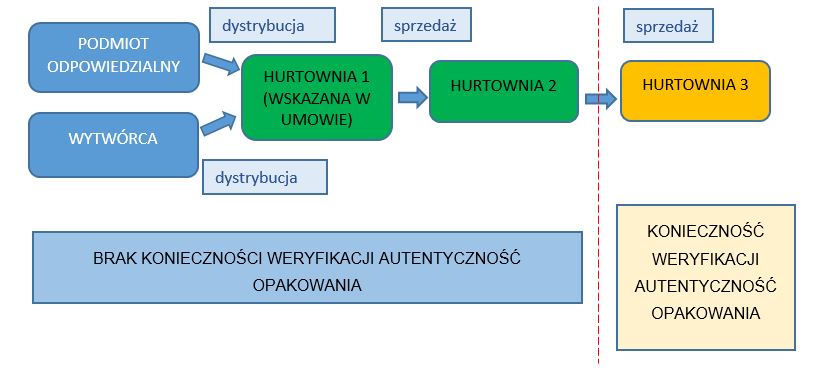
Jednak należy zauważyć, że w art. 21 Rozporządzenia Delegowanego Komisji (UE) 2016/161 z dnia 2 października 2015 r., wskazano następujące odstępstwa od konieczności przeprowadzania weryfikacji autentyczności niepowtarzalnego identyfikatora:
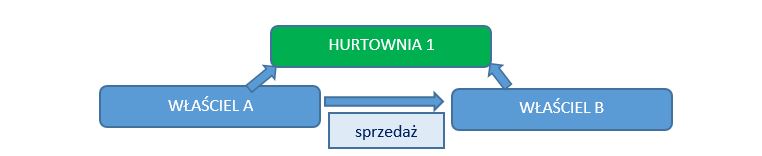
a) produkt leczniczy zmienia właściciela, lecz pozostaje w fizycznym posiadaniu tego samego hurtownika.
b) produkt leczniczy jest dystrybuowany na terytorium jednego państwa członkowskiego między dwoma magazynami należącymi do tego samego hurtownika lub do tego samego podmiotu prawnego, ale nie odbywa się sprzedaż.
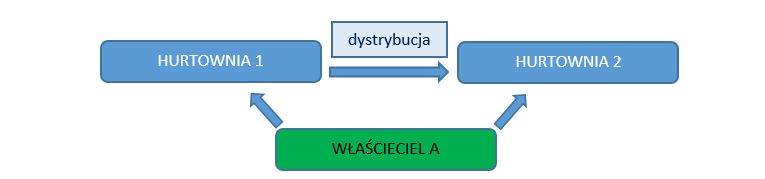
Powyższe schematy powinny zostać uwzględnione we wszystkich modelach dystrybucyjnych stosowanych przez przedsiębiorców posiadających zezwolenie na prowadzenie hurtowni farmaceutycznej.
- W jakich przypadkach weryfikacja niepowtarzalnego identyfikatora jest obowiązkowa?
Zgodnie z art. 22 Rozporządzenia Delegowanego Komisji Europejskiej nr 2016/161 hurtownia ma obowiązek wycofywać niepowtarzalny identyfikator w następujących przypadkach:
a) produktów, które zamierza dystrybuować poza Unię;
b) produktów, które zostały mu zwrócone przez osoby upoważnione lub uprawnione do dostarczania pacjentom produktów leczniczych lub przez innego hurtownika i nie mogą być zwrócone do zapasów przeznaczonych do sprzedaży;
c) produktów, które są przeznaczone do zniszczenia;
d) produktów będących w jego fizycznym posiadaniu, które są wymagane przez właściwe organy jako próbki;
e) produktów, które zamierza dystrybuować wśród osób lub instytucji, o których mowa w art. 78 ust. 1 pkt 3a ustawy Prawo Farmaceutyczne (Dz. U. z 2020 r. poz. 944 z późn. zm.).
- Czy umowa, o której mowa w art. 77a ustawy Prawo Farmaceutyczne może być zawarta pomiędzy podmiotem odpowiedzialnym a hurtowną, która kupuje produkty lecznicze od podmiotu odpowiedzialnego?
Umowa, o której mowa w art. 77a ustawy Prawo Farmaceutyczne może być zawarta pomiędzy podmiotem odpowiedzialnym a hurtownią, która kupuje produkty lecznicze od podmiotu odpowiedzialnego, ponieważ ww. artykuł nie określa, kto jest właścicielem leku. Zawarcie takiej umowy skutkuje uznaniem hurtowni za hurtownię desygnowaną, od której przyjęcie produktów leczniczych nie wymaga weryfikacji autentyczności.
- Kto powinien dokonywać w hurtowni farmaceutycznej weryfikacji zabezpieczeń oraz wycofywać niepowtarzalny identyfikator znajdujący się na opakowaniu?
Weryfikacja zabezpieczeń oraz wycofywanie niepowtarzalnego identyfikatora znajdującego się na opakowaniu powinny być wykonywane przez osobę wyznaczoną przez przedsiębiorcę. Potwierdzenie zlecenia wykonywania powyższych obowiązków powinno znaleźć się w zakresie obowiązków pracownika i w odpowiednich procedurach. Osoba wykonująca te czynności powinna uczestniczyć w szkoleniach dedykowanych temu zagadnieniu.
- Kiedy powinna odbywać się weryfikacja zabezpieczeń w przypadku zwrotów do hurtowni?
Weryfikacja zabezpieczeń powinna się odbyć w momencie przyjęcia zwrotu, przed wprowadzeniem do właściwej lokalizacji magazynu hurtowni. Takie postępowanie pozwoli na uniknięcie przyjęcia na stan hurtowni opakowania produktu leczniczego uprzednio wycofanego z systemu lub podejrzanego o sfałszowanie.
- W przypadku zwrotu produktu leczniczego przez aptekę do hurtowni farmaceutycznej z powodu pomyłki w zamówieniu lub błędu w dostawie, jaki status w systemie weryfikacji powinno posiadać opakowanie tego produktu?
W przypadku zwrotu produktu leczniczego przez aptekę do hurtowni farmaceutycznej z powodu pomyłki w zamówieniu lub błędu w dostawie rekomendujemy, aby apteka upewniła się, że zwracane opakowanie produktu posiada status „aktywne” w PLMVS.
- Czy czynności o których mowa w umowie zawartej zgodnie z art. 77a ustawy Prawo Farmaceutyczne mogę być podzlecane innym podmiotom?
Na podstawie art. 77a ust. 4 ustawy Prawo Farmaceutyczne (Dz. U. z 2020 r. poz. 944 z późn. zm.) przedsiębiorca prowadzący działalność polegającą na prowadzeniu hurtowni farmaceutycznej nie może zlecać podwykonawcom czynności określonych w umowie zawartej z podmiotem odpowiedzialnym o przechowywanie lub dostarczanie w imieniu podmiotu odpowiedzialnego produktu leczniczego, co do którego podmiot odpowiedzialny uzyskał pozwolenie na dopuszczenie do obrotu. Przepis ten wchodzi w życie 1 lipca 2020r.
- Ile hurtowni może być wyznaczonych w pisemnych umowach przez posiadacza pozwolenia na dopuszczenie do obrotu, do dystrybucji w jego imieniu produktów objętych pozwoleniem na dopuszczenie do obrotu?
Ustawa Prawo Farmaceutyczne oraz Rozporządzenie Delegowane Komisji Europejskiej nr 2016/161 (z dnia 2 października 2015 r. uzupełniającego Dyrektywę 2001/83/WE Parlamentu Europejskiego i Rady przez określenie szczegółowych zasad dotyczących zabezpieczeń umieszczanych na opakowaniach produktów leczniczych stosowanych u ludzi) nie wprowadzają ograniczeń w ilości hurtowni, które mogą być wyznaczone w pisemnych umowach przez posiadacza pozwolenia na dopuszczenie do obrotu, do dystrybucji w jego imieniu produktów objętych pozwoleniem na dopuszczenie do obrotu. Niemniej jednak należy mieć na uwadze, że odstępstwo od weryfikacji autentyczności opakowania, w przypadku hurtowni wyznaczonej w umowie, musi zostać uwzględnione w systemie zapewnienia jakości, w szczególności podczas kwalifikacji dostawców oraz stosownych procedurach i instrukcjach obowiązujących w hurtowni. Wykaz produktów leczniczych objętych ww. umową musi być dostępny dla osób wykonujących weryfikację autentyczności opakowań w hurtowni.
- Czy opakowania produktów leczniczych sprowadzone w ramach importu docelowego lub interwencyjnego podlegają obowiązkowi weryfikacji i wycofaniu zgodnie z rozporządzeniem delegowanym Komisji Europejskiej nr 2016/161?
Kwestia dotycząca obowiązku weryfikacji autentyczności opakowań produktów leczniczych wprowadzanych na terytorium państwa członkowskiego zgodnie z art 5 ust. 1 Dyrektywy 2001/83/WE Parlamentu Europejskiego i Rady (import docelowy) poruszona została w dokumencie Komisji Europejskiej „SAFETY FEATURES FOR MEDICINAL PRODUCTS FOR HUMAN USE, QUESTIONS AND ANSWERS – VERSION 17” pkt 1.8, w którym stwierdzono, że jeśli produkt leczniczy, zgodnie z art. 5 ust. 1 ww. Dyrektywy 2001/83/WE, jest wprowadzany na terytorium państwa członkowskiego zasady dotyczące zabezpieczeń wynikających z serializacji zasadniczo nie mają zastosowania. W związku z czym, wobec braku przepisów krajowych stanowiących inaczej, przepisy dotyczące zabezpieczeń nie mają zastosowania. Wspomniany powyżej pkt. 1.8 można traktować, jako przesłankę potwierdzającą możliwość wyłączenia stosowania obowiązków wynikających z Rozporządzenia Delegowanego Komisji 2016/161 również w przypadku, o którym mowa w art. 5 ust. 2 Dyrektywy 2001/83/WE, w czyli do produktów sprowadzanych do Polski w ramach importu interwencyjnego na podstawie art. 4 ust. 8 ustawy Prawo Farmaceutyczne (Dz. U. z 2020 r. poz. 994 z późn. zm.).
-
Na kim spoczywa obowiązek wycofania niepowtarzalnego identyfikatora z PLMVS w przypadku dystrybucji produktów leczniczych ze szpitala do pacjenta przez hurtownie farmaceutyczne w ramach Leczenia szpitalnego - programy lekowe oraz Leczenia szpitalnego - chemioterapia w okresie stanu zagrożenia epidemicznego lub stanu epidemii?
Zgodnie z Komunikatem Głównego Inspektora Farmaceutycznego z dnia 03 kwietnia 2020 r. istnieje możliwość dostarczenia produktów leczniczych przez hurtownię farmaceutyczną z apteki szpitalnej do pacjenta jedynie w ramach Leczenia szpitalnego - programy lekowe oraz Leczenia szpitalnego - chemioterapia w okresie stanu zagrożenia epidemicznego lub stanu epidemii. Wymagane jest, aby produkt leczniczy był wydawany zleceniobiorcy realizującemu usługę transportową przez aptekę szpitalną. Zgodnie z art. 25 Rozporządzenia Delegowanego Komisji (UE) 2016/161 z dnia 2 października 2015 r., do obowiązków osób upoważnionych lub uprawnionych do dostarczania pacjentom produktów leczniczych należy weryfikacja zabezpieczenia i wycofania niepowtarzalnego identyfikatora każdego produktu leczniczego zawierającego zabezpieczenia dostarczanego przez nich pacjentom w momencie dostarczenia go pacjentowi. Ponadto osoby upoważnione lub uprawnione do dostarczania pacjentom produktów leczniczych działające w instytucji opieki zdrowotnej mogą przeprowadzać weryfikację i wycofanie w dowolnym momencie, kiedy produkt leczniczy jest w fizycznym posiadaniu instytucji opieki zdrowotnej, pod warunkiem, że między dostarczeniem produktu leczniczego instytucji opieki zdrowotnej a dostarczeniem go pacjentowi nie ma miejsca jego sprzedaż. W związku z powyższym, wycofanie niepowtarzalnego identyfikatora produktu leczniczego przeznaczonego dla pacjentów w ramach Leczenia szpitalnego - programy lekowe oraz Leczenia szpitalnego - chemioterapia w okresie stanu zagrożenia epidemicznego lub stanu epidemii należy do obowiązku apteki szpitalnej.
- Czy zasady dotyczące zabezpieczeń mają zastosowanie do produktów leczniczych przeznaczonych do badań i testów rozwojowych?
Produkty lecznicze przeznaczone do badań naukowych i rozwojowych, którym nie przyznano jeszcze pozwolenia na dopuszczenie do obrotu, są wyłączone z przepisów dotyczących zabezpieczeń. Produkty lecznicze dopuszczone do obrotu muszą spełniać wymogi dyrektywy 2001/83/EC i Rozporządzenia Delegowanego Komisji (UE) 2016/161 do momentu, gdy wiadomo, która partia/jednostka zostanie wykorzystana w badaniach i testach rozwojowych.
W praktyce istnieją dwie możliwe sytuacje:
a) Produkt jest wytwarzany do zastosowania w badaniu klinicznym.
Badany produkt leczniczy (IMP), który jest wytwarzany zgodnie z pozwoleniem na dopuszczenie do obrotu, ale pakowany w celu przeprowadzenia badania klinicznego (nie występujący w postaci handlowej), jest wyłączony z przepisów dotyczących zabezpieczeń, ponieważ jest wytwarzany i pakowany wyłącznie w celu zastosowania w badaniu klinicznym. Wytwórca byłby zobowiązany do posiadania pozwolenia na wytwarzanie i import IMP i certyfikat IMP wydany na podstawie tego pozwolenia zgodnie z wnioskiem o badanie kliniczne, z zastrzeżeniem, że wniosek o badanie kliniczne musi odzwierciedlać te ustalenia. Referencyjne/pomocnicze produkty lecznicze dopuszczone do obrotu nie mogą być wytwarzane na podstawie pozwolenia na wytwarzanie i przywóz obejmującego IMP i muszą spełniać wymagania dotyczące opakowania wprowadzonego do obrotu, posiadającego cechy zabezpieczające i odpowiednio wycofanego z obrotu (zob. poniżej).
b) Produkt jest dopuszczony do obrotu i pochodzi z łańcucha dostaw.
Produkty lecznicze w ich postaciach zgodnej z pozwoleniem na dopuszczenie do obrotu posiadające zabezpieczenia, powinny być wycofane z systemu weryfikacji autentyczności, przed ich zastosowaniem jako badanych produktów leczniczych lub pomocniczych produktów leczniczych dopuszczonych do obrotu.
- Czy istnieje możliwość dostarczenia leku do ośrodka prowadzącego badania kliniczne i wydania pacjentowi leku bez potwierdzenia autentyczności oraz wycofania identyfikatora?
Nie, do obowiązków osób upoważnionych lub uprawnionych do dostarczania pacjentom produktów leczniczych należy weryfikacja zabezpieczenia i wycofania niepowtarzalnego identyfikatora każdego produktu leczniczego zawierającego zabezpieczenia dostarczanego przez nich pacjentom w momencie dostarczenia go pacjentowi. Zatem w przypadku prowadzenia badania klinicznego w zakładach leczniczych weryfikacja i wycofanie powinno zostać przeprowadzone w aptece szpitalnej lub dziale farmacji szpitalnej. Należy zauważyć, że zgodnie z art. 78 ust.1, pkt 3a ustawy Prawo farmaceutyczne (Dz. U. z 2020 r. poz. 944 z póź. zm.), do obowiązków przedsiębiorcy prowadzącego działalność polegającą na prowadzeniu hurtowni farmaceutycznej należy weryfikowanie zabezpieczeń, o których mowa w art. 54 lit. o Dyrektywy 2001/83/WE, i wycofanie niepowtarzalnego identyfikatora produktu leczniczego, o którym mowa w art. 3 ust. 2 lit. a rozporządzenia nr 2016/161, przed dostarczeniem tego produktu m.in. dla:
- lekarzy, pielęgniarek i położnych wykonujących działalność leczniczą w formie praktyki zawodowej, o której mowa w art. 5 ustawy z dnia 15 kwietnia 2011 r. o działalności leczniczej,
- aptek zakładowych, o których mowa w art. 87 ust. 1 pkt 3 ustawy Prawo Farmaceutyczne.
- Czy w przypadku braku podłączenia do PLMVS podmiot wydający produkt leczniczy pacjentowi może dokonać późniejszej weryfikacji autentyczności opakowania, po uzyskaniu z Fundacji KOWAL certyfikatu do systemu?
Weryfikacja i wycofanie produktu leczniczego z krajowego systemu weryfikacji autentyczności leków powinno nastąpić zanim produkt leczniczy trafi do pacjenta. Tylko taki sposób postępowania gwarantuje, że pacjent nie dostanie leku sfałszowanego.
- Jak powinna postąpić osoba upoważniona lub uprawniona do dostarczania pacjentom produktów leczniczych w przypadku wystąpienia problemu technicznego uniemożliwiającego weryfikację autentyczności niepowtarzalnego identyfikatora np. braku prądu, zerwania połączenia z internetem, awaria oprogramowania?
Zgodnie z art. 29 rozporządzenia delegowanego 2016/161, jeżeli problem techniczny uniemożliwia osobom upoważnionym lub uprawnionym do dostarczania pacjentom produktów leczniczych weryfikację autentyczności i wycofanie niepowtarzalnego identyfikatora w momencie dostarczenia pacjentowi produktu leczniczego opatrzonego tym identyfikatorem, osoby upoważnione lub uprawnione do dostarczania pacjentom produktów leczniczych odnotowują niepowtarzalny identyfikator i, jak tylko problemy techniczne zostaną rozwiązane, weryfikują autentyczność niepowtarzalnego identyfikatora i wycofują go.
- Czy podmiot prowadzący działalność leczniczą, nieposiadający apteki szpitalnej, powinien mieć dostęp do systemu PLMVS?
Podmioty lecznicze wykonujące działalność leczniczą w świetle ustawy z dnia 15 kwietnia 2011 r. o działalności leczniczej (Dz. U. Nr 112, poz. 654 ), posiadające księgi rejestrowe podmiotów leczniczych, nie są zwolnione z obowiązku serializacji wynikającego z Rozporządzenia Delegowanego Komisji UE 2016/161 z dnia 2 października 2015 roku uzupełniającego dyrektywę 2001/83/WE Parlamentu Europejskiego i Rady przez określenie szczegółowych zasad dotyczących zabezpieczeń umieszczanych na opakowaniach produktów leczniczych stosowanych u ludzi.
Ponadto, zgodnie z art. 87 ust. 4 ustawy Prawo Farmaceutyczne w zakładach leczniczych podmiotów leczniczych, w których nie utworzono apteki szpitalnej albo apteki zakładowej, tworzy się dział farmacji szpitalnej, do zadań, którego należy m. in.: wydawanie produktów leczniczych i wyrobów medycznych określonych w odrębnych przepisach oraz ustalanie procedur wydawania produktów leczniczych lub wyrobów medycznych przez aptekę szpitalną na oddziały oraz dla pacjenta.
W związku z powyższym dostęp do PLMVS powinien posiadać działy farmacji szpitalnej bądź apteki szpitalne funkcjonujące w zakładach leczniczych.
- Czy każda hurtownia, apteka ogólnodostępna, apteka szpitalna czy dział farmacji szpitalnej powinny posiadać indywidualny dostęp do systemu?
Każda hurtownia, apteka ogólnodostępna apteka szpitalna oraz dział farmacji szpitalnej, powinny mieć indywidualny dostęp do systemu, który umożliwia weryfikację i wycofanie opakowania z systemu w miejscu, w którym zostało ono faktycznie wydane. W przypadku korzystania z jednego certyfikatu w różnych miejscach weryfikacja użytkownika dokonującego wycofania opakowania z systemu jest niemożliwa do wykonania.
- Jakie są najczęstsze problemy techniczne po stronie detalicznych użytkowników końcowych PLMVS (apteki ogólnodostępne, apteki szpitalne, działy farmacji szpitalnej)?
Najczęstszymi błędami technicznymi są nieprawidłowa konfiguracja czytnika kodu 2D Data Matrix (skaner) lub błędy oprogramowania prowadzące do nieprawidłowej interpretacji zawartości kodu 2D Data Matrix. W obydwu przypadkach błędy prowadzą do przesłania do bazy PLMVS informacji niezgodnych z zawartością kodu 2D Data Matrix opakowania leku, co prowadzi do pojawienia się alertu, o którym mowa w art. 36(b) Rozporządzenia Delegowanego Komisji (UE) 2016/161.
- W jaki sposób uniknąć błędu nieprawidłowej konfiguracji czytnika kodu 2D Data Matrix (skanera)?
Należy skontaktować się ze swoim opiekunem serwisowym po stronie dostawcy oprogramowania aptecznego lub dostawcy skanera w celu uzyskania pomocy technicznej. Większość aptek posiada umowy serwisowe, z których należy korzystać. Niektóre z systemów aptecznych posiadają również moduły diagnostyczne pozwalające na samodzielną weryfikację prawidłowości konfiguracji. Najprostszym sposobem na zdiagnozowanie błędu konfiguracji powodującego zamianę liter wielkich na małe w numerach serii (partii) lub numerach seryjnych opakowania leku jest weryfikacja leku z użyciem skanera, a w przypadku pojawienia się alertu, powtórna weryfikacja po zwolnieniu klawisza „CapsLock” lub ”Caps”. Jeżeli po zwolnieniu klawisza CapsLock (włączany i wyłączany poprzez jednokrotne wciśnięcie) alert nie pojawia się, oznacza to, że oprogramowanie apteczne jest wrażliwe na przycisk CapsLock. W takiej sytuacji, albo należy powstrzymać się przed korzystaniem z przycisku CapsLock (zamiana liter na wielkie na klawiaturze), albo zwrócić się do dostawcy oprogramowania aptecznego o aktualizację oprogramowania lub konfigurację skanera, stosownie do potrzeb użytkownika aptecznego. Zazwyczaj można tego dokonać w ramach zgłoszeń serwisowych u dostawców oprogramowania aptecznego lub dostawców skanera dla apteki. Alternatywną przyczyną błędów w odczycie kodów 2D Data Matrix może być błąd oprogramowania aptecznego. W tym przypadku zachęcamy do zapoznania się z odpowiedzią na pytanie pt.: „W jaki sposób uniknąć błędów oprogramowania aptecznego w komunikacji z PLMVS?”.
- W jaki sposób uniknąć błędów oprogramowania aptecznego w komunikacji z PLMVS?
Zalecanym rozwiązaniem dla uniknięcia błędów oprogramowania aptecznego w odczycie kodów 2D Data Matrix jest skorzystanie przez dostawcę IT apteki z bezpłatnych testów jakości oprogramowania służącego weryfikacji autentyczności leków (tzw. Baseline Testing). Dostęp do narzędzi weryfikujących prawidłowość działania oprogramowania aptecznego dostępny jest dla każdego dostawcy IT oferującego narzędzia informatyczne służące weryfikacji autentyczności leków pod adresem: https://sws-nmvs.eu/?destination=baselineTesting. W razie zdiagnozowania w aptece ogólnodostępnej, aptece szpitalnej lub dziale farmacji szpitalnej dużej liczby alertów w codziennej pracy apteki, wysoce prawdopodobne jest, że oprogramowanie apteczne nie przeszło testów jakości oprogramowania i nieprawidłowo interpretuje zawartość kodów 2D Data Matrix na opakowaniu leku. Zalecamy kontakt z dostawcą IT w celu potwierdzenia pomyślnego przeprowadzenia testów jakości oprogramowania (Baseline Testing), a jeśli nie miały miejsca, wyrażenie oczekiwania ich przeprowadzenia. Alternatywną przyczyną błędów w odczycie kodów 2D Data Matrix może być błąd konfiguracji czytnika. W tym przypadku zachęcamy do zapoznania się z odpowiedzią na pytanie pt.: „W jaki sposób uniknąć błędu nieprawidłowej konfiguracji czytnika kodu 2D Data Matrix (skanera)?”.